Un phéochromocytome est un type rare de tumeur dont les symptômes peuvent ressembler à l’hypertension, à l’hypoglycémie, à des problèmes de thyroïde ou même à une crise de panique. Voilà certains des symptômes dont Bryde Fresque a fait l’expérience. Lisez son histoire incroyable.
L’Hôpital d’Ottawa est à l’avant-garde de la recherche et des traitements du phéochromocytome grâce à de nombreux experts en la matière.
Communément appelée « phéo », la tumeur n’est pas décelée dans de nombreux cas, car ses symptômes – hypertension, battement rapide du cœur et transpiration – sont épisodiques et soudains.
Ces symptômes sont causés par des phéochromocytomes qui se forment habituellement dans l’une des glandes surrénales du corps et entraînent une production excessive d’adrénaline. Les histoires des patients sont parfois très dramatiques et racontent des symptômes mystérieux ou une insuffisance cardiaque soudaine chez des jeunes, autrement en bonne santé.
Pourquoi le phéochromocytome est-il rare?
On décèle un ou deux cas de phéochromocytome sur 100 000 personnes. La tumeur touche autant les hommes que les femmes de tous âges, quoique plus fréquente chez les adultes âgés de 30 à 50 ans. La plupart des cas surviennent de façon aléatoire et mystérieuse et environ le tiers sont d’origine génétique héréditaire.
Le saviez-vous? Le cas du président Dwight D. Eisenhower est un exemple célèbre. C’est après sa mort qu’on a découvert qu’un phéochromocytome avait peut-être contribué à son hypertension.
Comment diagnostique-t-on le phéochromocytome?
Les médecins recherchent un phéochromocytome lorsque des patients autrement en bonne santé ont des symptômes inexplicables. Pour ce faire, un médecin demandera des analyses d’urine et de sang pour vérifier les taux d’hormone et utilisera la tomodensitométrie pour déceler la présence d’une tumeur.
Comment traite-t-on le phéochromocytome?
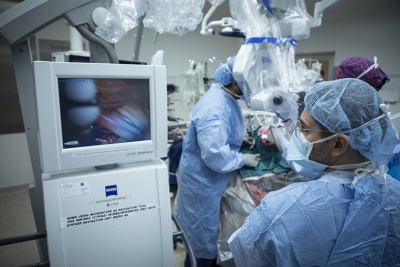
La chirurgie est le traitement habituel pour retirer complètement la glande surrénale. L’intervention peut s’avérer dangereuse, car elle stimule la tumeur et peut entraîner l’augmentation de la tension artérielle. Pour éviter cela, les médecins, les endocrinologues et les anesthésiologistes travaillent ensemble pour contrôler la tension artérielle avant et pendant la chirurgie.

« Après la chirurgie, j’ai reçu mon diagnostic, qui expliquait mes symptômes antérieurs. J’avais un phéochromocytome – une tumeur rare potentiellement cancéreuse. »
— Bryde Fresque
Le phéochromocytome est-il un cancer?
Le phéochromocytome peut être cancéreux, mais la plupart ne le sont pas. Environ 10 % seulement des phéochromocytomes se propagent à d’autres organes. Néanmoins, on recommande de le retirer à cause du risque qu’il pose. Sans traitement, un phéochromocytome peut entraîner une insuffisance cardiaque, un AVC et une insuffisance rénale.
Pour un phéochromocytome cancéreux, d’autres traitements peuvent inclure la radiothérapie, la chimiothérapie ou des traitements ciblés contre le cancer pour empêcher la tumeur de grandir et de se propager.
Que réserve la recherche sur le phéochromocytome?
Actuellement, la plupart des études sur le phéochromocytome portent sur ses causes génétiques. En effet, plusieurs gènes qui contribuent à son apparition sont déjà connus. Les chercheurs espèrent pouvoir déceler tôt les phéochromocytomes, identifier les personnes à risque et mieux comprendre la formation et le traitement de la tumeur.
L’Hôpital d’Ottawa est un chef de file non seulement dans le traitement des phéochromocytomes, mais aussi dans la recherche sur les maladies rares comme les tumeurs surrénales « phéo ». Cette combinaison rend possibles des soins de calibre mondial, ici même à Ottawa.